Canalopathie lié à KCNMA1
This French translation of our English version is courtesy of Dr. Armand Thibaud, Pediatrician, Dept of Clinical Genetics, CHU de Lyon HCL - GH Est-Hôpital Femme Mère Enfant.
[Le texte ci-dessous est une vulgarisation scientifique destinée aux patients et à leur famille. Il existe une revue publiée complète de la canalopathie liée à KCNMA1, rédigée par la communauté scientifique et médicale en annexe.]
QU'EST-CE QU'UNE CANALOPATHIE?
Un «canal» est pour simplifier un tunnel qui permet à un ou plusieurs «ions» spécifiques (atome portant une charge électrique : telles que le sodium, le potassium, le chlorure, etc.) de traverser une membrane, mais uniquement dans des conditions très spécifiques.
Les cellules qui composent notre corps contiennent à leur surface de nombreux types de canaux ioniques qui permettent aux ions d'entrer et de sortir des cellules. Le flux d'ions est étroitement régulé et est essentiel pour assurer et maintenir le bon fonctionnement de notre organisme. Ce flux permet de multiples activités physiologique, tels que le contrôle de l'activité électrique du cerveau, des muscles et du cœur. Lorsque l’activité de ces canaux ioniques est modifiée, les individus peuvent développer des symptômes associés à un large éventail de troubles appelés «canalopathies».
Le type de symptômes présentés par un patient dépend du type de canal ionique affecté, car différents canaux ioniques remplissent des fonctions différentes et sont exprimés à différents endroits dans le corps.
Les canalopathies sont généralement le résultat de défauts génétiques (mutations). Cependant, certaines canalopathies peuvent être causées par des médicaments, des drogues, des poisons ou des maladies auto-immunes.
QU'EST-CE QUE LA CANALOPATHIE LIÉE À KCNMA1 ?
Il s'agit d'une pathologie dans lequel la majorité des patients peuvent avoir des crises d'épilepsie, des mouvements anormaux involontaires, ou les deux. D'autres symptômes peuvent également survenir. Elle est causée par certaines mutations d'un gène appelé KCNMA1. La partie "KCN" du nom est l'abréviation de "Canal Potassique". “K” est l'abréviation de potassium dans le tableau périodique des éléments. La partie «MA1» est le nom du type spécifique de gène et de canal du potassium affectés. Par exemple, d'autres troubles sont provoqués par des mutations de KCNA1, KCNQ2, KCNQ3, KCNJ2, etc. Nous résumons ici un examen complet de la canalopathie liée à KCNMA1.
QUELS AUTRES CONDITIONS MÉDICALES ET SYMPTÔMES SONT ASSOCIÉS À LA CANALOPATHIE LIÉE À KCNMA1?
La canalopathie liée à KCNMA1 est un syndrome, c'est-à-dire une combinaison de symptômes qui se produisent ensemble. Il existe une grande variation dans le nombre et la gravité des symptômes ressentis par les patients atteints de cette maladie. De nombreuses personnes atteintes de ce syndrome présentent un trouble du mouvement, parfois appelé «dyskinésie paroxystique non kinésigénique». D'autres patients peuvent avoir des crises d'épilepsie de divers types. Certains peuvent avoir des troubles du mouvement et des crises d'épilepsie. Beaucoup ont également des retards de développement et d'autres symptômes neurologiques.

DYSKINESIE PAROXYSTIQUE NON KINESIGENIQUE (PNKD) TYPE 3
L'un des symptômes les plus courants chez les personnes atteintes de certaines mutations de KCNMA1 est constitué par des mouvements involontaires très soudains, brusques. La «dyskinésie» est un terme médical qui signifie simplement des mouvements anormaux involontaires. Le terme «paroxystique» signifie qu’il peut commencer très rapidement et que les symptômes vont et viennent, avec des périodes asymptomatiques entre les deux. Enfin, «non kinésigénique» signifie que les mouvements anormaux ne sont pas «déclenchés» par certains mouvements particuliers, comme cela peut se produire dans d’autres formes de dyskinésie. Il existe au moins trois gènes différents associés à ce type de trouble du mouvement. Des mutations dans KCNMA1 ont été les troisièmes à être découvertes, c'est pourquoi on l'appelle parfois PNKD « Type 3 ».
Les crises de dyskinésie peuvent durer de quelques secondes à quelques minutes avant de s’arrêter. Certains patients peuvent avoir plusieurs dizaines d'attaques par jour. Les gens se déplacent normalement entre les attaques. Les attaques dyskinétiques musculaires peuvent prendre de nombreuses formes et consister en une torsion, un raidissement ou une mauvaise coordination d’une ou de plusieurs parties du corps, voire du corps tout entier. Dans certains cas, le patient peut ne plus être dans la capacité de bouger, de parler ou de réagir. Cependant, les gens sont conscients et éveillé. Étant donné que les mouvements peuvent être protéiformes selon les personnes, un nom plus spécifique leur est parfois attribué en fonction de la forme particulière de mouvement anormal observée chez cette personne. Ceux-ci incluent: chorée, atonie, dystonie et autres.
DROP ATTACK / CHUTE BRUSQUE PAR DEROBEMENT DES JAMBES
Un autre symptôme caractéristique chez certains patients atteints de canalopathie liée à KCNMA1 est appelé « drop attack ». Au cours de ces épisodes, les patients perdent soudainement le contrôle de leurs muscles et peuvent tomber au sol sans avertissement. Dans une « drop attack », les muscles des bras et des jambes peuvent être soit tendus, soit complètement mous, ces crises surviennent cependant toujours involontairement. D'autres termes parfois utilisés pour les «drop attack», sont atonie ou akinésie. Parfois, la bouche ou la langue peuvent faire des mouvements étranges lors d’une « drop attack ». La durée de ces attaques peut durer jusqu'à 20 secondes et peut se produire plusieurs fois par jour. Les « drop attack », sont souvent confondus avec des crises d'épilepsie car certaines crises d’épilepsie peuvent avoir une présentation très proche. Notez que le terme « drop attack », peut prêter à confusion, car il signifie différentes choses selon le contexte et le sens médical de ce terme n’est pas bien défini.
ÉPILEPSIE
Beaucoup d'enfants et d'adultes atteints de canalopathie liée à KCNMA1 ont une épilepsie. L'épilepsie est un symptôme neurologique causé par un dysfonctionnement passager du cerveau ; certains disent qu'il « court-circuite ». Lors d'une crise d'épilepsie, les neurones (cellules nerveuses cérébrales) produisent soudainement une décharge électrique anormale dans certaines zones du cerveau. En général, les crises chez les personnes atteintes d'un trouble lié à KCNMA1 impliquent le « cerveau entier », ce que les spécialistes qualifient d'épilepsie « généralisée ». Il existe de nombreux types de crises d'épilepsie possibles. Certaines crises d'épilepsie consistent uniquement en de très brèves secousses musculaires (« saccades myocloniques » ou crises « myocloniques), à des convulsions avec raideur du corps entier et perte de conscience (crises « toniques »), à des convulsions avec mouvements saccadés rythmiques soutenus brefs associés à une raideur musculaire («crises cloniques»), ou à des crises où le patient perd conscience de manière brève, ne répondent pas et ne peuvent pas voir ni entendre sans perturbation du tonus musculaire («absence»). Les médecins peuvent décrire les crises par ce qui se passe à l’extérieur (ce que l’on peut « voir », la clinique) ou parfois par ce qui se passe à l’intérieur (par exemple, l’aspect du tracé de l’électroencéphalogramme) ou les deux (on parle alors de description électro-clinique). Les patients atteints de canalopathie liée à KCNMA1 peuvent ne pas présenter de crise. A l’inverse, plusieurs types de crises peuvent se produire en même temps ou les unes après les autres. Les crises peuvent commencer à n'importe quel âge.

RETARD DE DÉVELOPPEMENT
Les enfants atteints de canalopathie liée à KCNMA1 peuvent avoir un développement et une intelligence normale. Ils peuvent apprendre à s'asseoir, se tenir debout, marcher et parler en même temps que les autres enfants. Mais généralement, les enfants souffrant de crises d'épilepsie fréquentes ont tendance à se développer plus lentement que les autres enfants on parle alors de retard de développement psychomoteur.
QUELS AUTRES PROBLÈMES POUR LES PERSONNES ATTEINTES D'UN TROUBLE LIÉ À KCNMA1 ?
Comme nous connaissons peu de patients atteints de cette maladie (environ 50 seulement !), Il est fort possible que d’autres symptômes ne soient pas encore décrits. Toute personne chez laquelle une canalopathie liée à KCNMA1 a été diagnostiquée est encouragée à contacter la fondation « KCNMA1 Channelopathy International Advocacy Foundation » afin d’aider les neurologues et les scientifiques à mieux définir les symptômes et les réponses aux thérapies usuelles.
QUELLE EST LA GRAVITÉ D'UNE ATTEINTE LIÉE À KCNMA1 ?
De nombreuses personnes ayant une mutation KCNMA1 présentent des symptômes minimes voir absents. En contrepartie certains patients ont de graves problèmes qui nuisent directement à leur santé, un large panel d’expression de la maladie est retrouvé chez les patients entre ces deux extrêmes.
Il n’y a pas de corrélations entre le type de mutation est la sévérité de la maladie sur le plan clinique et des personnes ayant une même mutation peuvent tout à fait avoir des symptômes très différents. En outre, les symptômes peuvent évoluer avec le temps chez une même personne, avec des symptômes pouvant s’améliorer ou s’aggraver.

QUELLE EST LA FREQUENCE DE LA CANALOPATHIE LIEE A KCNMA1 ?
C'est une maladie très rare. On estime que moins de 1 personne sur 100 000 est touchée. Mais grâce à l’émancipation des nouveaux outils de génétique moléculaire, les patients sont de plus en plus diagnostiqués.
CHEZ QUI LA CANALOPATHIE LIÉE À KCNMA1 SURVIENT ?
Comme il s’agit d’une mutation génétique, la canalopathie liée à KCNMA1 est déjà présente avant la naissance, même si les symptômes peuvent ne se manifester que beaucoup plus tard.
QU'EST-CE QUI CAUSE UNE CANALOPATHIE LIÉE À KCNMA1 ?
Elle est causée par une mutation d'un gène appelé KCNMA1, il se situe sur le chromosome 10. Ce gène est responsable de la fabrication de l'une des centaines de protéines différentes qui assurent le bon fonctionnement de l'activité électrique des cellules du cerveau, des nerfs, du cœur et des muscles. C'est pourquoi bon nombre des symptômes concernent le cerveau et les muscles. Différents types de mutations de KCNMA1 ont été identifiés chez des patients atteints de cette affection et chaque mutation peut entraîner un ensemble unique de symptômes. Il est également possible que des patients partageant le même type de mutation KCNMA1 présentent des symptômes différents.

MUTATION KCNMA1 de novo
À l’heure actuelle, la plupart des canalopathies liées à KCNMA1 présentent des mutations «de novo», ce qui signifie que la mutation du gène KCNMA1 s’est produite de manière aléatoire dans un ovule ou un spermatozoïde, mais n’a été transmise par aucun des parents.

AUTOSOMIQUE DOMINANT (HETEROZYGOTE) VS. AUTOSOMIQUE RECESSIF (HOMOZYGOTE)
La canalopathie liée à KCNMA1 est le plus souvent considérée comme une mutation « autosomique dominante » ou hétérozygote, c’est-à-dire qu’elle nécessite uniquement qu’il existe un problème dans l’un des deux gènes KCNMA1 que nous avons tous. En revanche, une mutation « autosomique récessive » ou homozygote nécessite que les deux copies de ce gène portent une mutation afin de provoquer des symptômes. Seules quelques personnes atteintes d'une affection KCNMA1 récessive sont connues.
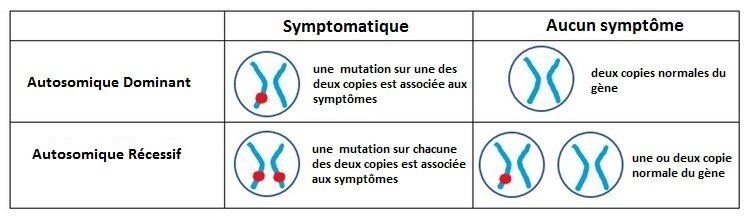
UNE MUTATION KCNMA1 PEUT-ELLE ÊTRE HÉRITÉE?
Chez quelques patiente, la mutation de KCNMA1 était héritée d'un parent. Les enfants d'un adulte avec la forme dominante de cette maladie ont 50% de chances d'hériter du gène mutant. Cependant, bien qu’il puisse avoir la même mutation, il n’est pas possible de prédire si les symptômes de l’enfant seront identiques à ceux de ses parents. Les symptômes peuvent être similaires, plus légers ou plus graves. Chez ceux qui ont la forme autosomique récessive ou homozygote, le risque que leurs propores enfants soit atteint est très faible (il faudrait que le conjoint soit aussi porteur), ce qui est peu probable compte tenu de la rareté des mutations de KCNMA1). Mais si les deux parents avaient effectivement des mutations KCNMA1, chaque enfant aurait 25% de chances d'hériter des deux gènes KCNMA1 anormaux. Pour plus d'informations sur la génétique et les modes de transmission génétique, veuillez visiter cette page.

PÉNÉTRANCE INCOMPLÈTE DES SYMPTÔMES
Le terme « pénétrance » fait référence au degré de symptômes qu’un patient présente pour une mutation génétique donnée. Toutes les personnes qui ont une erreur dans le gène KCNMA1 ne présentent pas de symptômes. Certaines personnes portant une mutation KCNMA1 peuvent ne jamais présenter de symptômes au cours de leur vie. Ce phénomène est appelé pénétrance incomplète.
QUE FAIT LE GENE KCNMA1?
Le gène KCNMA1 contient des informations pour la construction d'un type spécifique de protéine nécessaire aux cellules du corps. Cette protéine est nécessaire pour garantir le bon fonctionnement des signaux électriques dans les cellules, notamment le cerveau, le cœur et les muscles. La protéine codée par KCNMA1 est un «canal ionique», qui est un type de tunnel qui permet aux ions de circuler à travers les membranes cellulaires. Plus précisément, KCNMA1 constitue un type de canal unique qui permet aux ions potassium (K +) de circuler à travers le canal. Le canal KCNMA1 a de nombreux noms et est souvent appelé canal «BK» («Big K +»). Les canaux BK ont des propriétés spéciales, en ce sens qu’ils deviennent actifs lorsqu’un changement de tension est détecté alors qu’un ion (calcium) différent « touche » ou lie simultanément le canal. En raison de ce type d'activation unique, les canaux KCNMA1 jouent un rôle important dans l'activité électrique des cellules cérébrales et musculaires. Les mutations de KCNMA1 qui font que ce canal ionique fonctionne anormalement peuvent à leur tour conduire à une activité électrique anormale dans le cerveau ou les muscles. C'est pourquoi des problèmes dans n'importe lequel des nombreux types de canaux potassiques peuvent causer des crises d’épilepsie et des troubles du mouvement.
QUELLE EST LA FONCTION D'UN CANAL DE POTASSIUM?
La principale fonction des canaux potassiques est de contrôler le mouvement des ions potassium dans ou hors des cellules. Ce faisant, ces canaux aident à régler le voltage à travers la membrane d’une cellule et modulent la communication électrique entre les cellules. Lorsqu'ils sont activés, la plupart des canaux potassiques (y compris le type BK fabriqué par KCNMA1) permettent aux ions potassium de sortir de la cellule, ce qui rend le voltage plus négatif ; ce qui stabilise la cellule. D'une certaine manière, les canaux potassiques agissent comme un « frein » pour empêcher la cellule d'être active au mauvais moment. Certaines mutations de KCNMA1, font que le canal potassique BK ne fonctionne pas correctement, ce qui rend la cellule plus excitable. A l'inverse, d’autres mutations diminuent de façon plus importante son excitabilité. Ces effets au niveau cellulaire ont pour conséquence d’affecter l'équilibre global de la communication dans le cerveau, les nerfs ou les muscles, ce qui peut entraîner les nombreux symptômes observés dans les affections liées à KCNMA1.

QUE SIGNIFIE KCN DE KCNMA1?
« KCN » est l’acronyme donné aux gènes qui codent pour les différents types de canaux potassiques (K +) présents dans l’ensemble du corps. Toute « classe » ou type de canal potassique peut contrôler le mouvement des ions potassium dans ou hors des cellules. Mais les conditions dans lesquelles les canaux sont actifs / ouverts peuvent être très différentes selon la classe de canal. Les symboles qui suivent KCN fournissent plus de détails sur les caractéristiques spécialisées d'une classe particulière de canaux potassiques. Par exemple, les gènes « KCNM » et « KCNN » codent pour des canaux « activés par le calcium » du potassium (c’est-à-dire BK), tandis que les gènes « KCNT » codent pour des canaux « activés par le sodium » du potassium. À ce jour, 79 gènes KCN ont été identifiés et beaucoup ont été liés à des canalopathies causant des maladies. Voir cette page pour plus d’informations sur les différents gènes « KCN ».
COMBIEN DE MUTATIONS ONT ÉTÉ TROUVÉES DANS KCNMA1?
Bien que les troubles du mouvement et l'épilepsie soient certainement des maladies très courantes, ce n'est que très récemment que nous avons appris que les mutations de KCNMA1 peuvent être à l'origine de ces symptômes. Quinze mutations causant des maladies ont été décrites et publiées à ce jour dans la littérature, et beaucoup d’autres ont été fournies par les rapports génétiques des patients. Nous connaissons actuellement environ 50 patients dans le monde porteurs de mutations de KCNMA1 et présentant des symptômes. Si vous souhaitez participer aux efforts visant à comprendre comment les mutations de KCNMA1 produisent des symptômes, contactez-nous.
COMMENT UNE CANALOPATHIE LIÉE À KCNMA1 EST-ELLE DIAGNOSTIQUÉE ?
Grossesse et naissance
Il n'y a généralement pas de signes d’appels pendant la grossesse. Les patients atteints de canalopathie liée à KCNMA1 ont généralement un poids et une taille de naissance normal, et les premiers mois de vie sont généralement normaux.
Histoire de la maladie et recherche
Si un patient souffre de crises épileptiques graves et / ou de mouvements involontaires débutant dans l’enfance, sans cause claire pour ces symptômes, et ne réagit pas aux traitements médicaux classiques, un neurologue suggérera probablement un test génétique. N'oubliez pas qu'il existe de nombreuses maladies pouvant provoquer des convulsions et des troubles du mouvement. Les patients atteints de canalopathie liée à KCNMA1 peuvent également avoir d'autres troubles non liés à leur mutation qui contribuent aux symptômes qu'ils ressentent.
Électroencéphalogramme (EEG)
Lorsque l'on pense que les patients souffrent d’épilepsie, le premier test diagnostique réalisé est un EEG. Ce test enregistre l’activité électrique du cerveau pour déterminer si une activité électrique anormale est responsable des symptômes de la personne. Les « anomalies épileptiformes généralisées » sont un type d'anomalie souvent observé sur l'EEG de patients présentant une canalopathie liée à KCNMA1. Parmi ceux présentant des anomalies EEG, on parle couramment de « complexes de pointes-ondes généralisées », avec une fréquence comprise entre 3 et 4 Hz. Il existe d’autres formes d’onde anormales détectées par l’EEG, qui varient en fonction de l’amplitude, de la fréquence, de la durée, du type de crise et de la localisation de la crise dans le cerveau. Gardez à l'esprit qu'un EEG normal n'exclut pas toujours le diagnostic de convulsions. En outre, des anomalies de l’EEG peuvent ne pas être se manifester par des crises d'épilepsie.
IRM cérébrale
Chez les enfants atteints d’épilepsie et / ou de troubles du mouvement, une IRM du cerveau est souvent réalisée pour déterminer si des anomalies cérébrales anatomiques peuvent contribuer aux symptômes du patient. Cependant, les IRM réalisés chez les patients présentant une canalopathie liée à KCNMA1 sont généralement normaux. En effet, le problème réside dans le fonctionnement des cellules cérébrales, et non pas dans leur organisation ni dans des modifications des structures anatomique cérébrales.
Test ADN
Si d'autres tests de diagnostic médical ne permettent pas de confirmer un diagnostic définitif en cas de convulsions ou de troubles du mouvement, des tests génétiques peuvent être effectués pour rechercher une mutation dans des gènes impliqués dans les épilepsies. Plusieurs tests génétiques sont disponibles, dénommés « panels de gènes impliqués dans les épilepsies ». Ils permettent de rechercher des mutations dans KCNMA1 ainsi que des centaines d’autres gènes des canaux ioniques liés à l’épilepsie et à des mouvements anormaux. Les mutations de KCNMA1 peuvent également être détectées à l’aide d’un test génétique appelé « séquençage de l’exome ». Pour obtenir un test génétique, un neurologue adressera le patient à un médecin généticien, qui prescrira le test et rendra les résultats. Les tests génétiques libre d’accès disponibles sur internet (à savoir 23andme© ou GeneSavvy©) ne permettent pas de diagnostiquer cette affection.
COMMENT LA CANALOPATHIE LIÉE À KCNMA1 EST-ELLE TRAITÉE ?
Actuellement, il n’existe pas de traitement spécifique pour les affections liées à KCNMA1. Chaque type de mutation entraîne un ensemble différent de symptômes et chaque type de mutation peut comporter différents traitements qui pourraient aider à atténuer l’un des nombreux symptômes que peuvent présenter les patients. En fait, les personnes ayant la même mutation peuvent avoir des symptômes différents et une réponse différente aux mêmes médicaments. Il est très important que les patients agissent en étroite collaboration avec leur neurologue pour rechercher les meilleures options de traitement en fonction de leurs symptômes et de leur situation globale.
Traitement de l'épilepsie
Les médicaments appelés médicaments antiépileptiques (AE) constituent généralement la première étape pour tenter de prévenir les crises. On sait beaucoup de choses sur les médicaments contre les crises convulsives qui pourraient convenir à un type de crise particulier. À ce stade, on en sait très peu sur toutes les diverses mutations de KCNMA1 et sur les médicaments qui pourraient le mieux fonctionner (ou ne pas fonctionner du tout) pour une mutation donnée. N'oubliez pas que chaque personne atteinte d'une maladie liée à KCNMA1 n'a pas nécessairement la même maladie ! Si les médicaments ne permettent pas de contrôler les crises d'épilepsie après en avoir essayé plusieurs, il existe d'autres options thérapeutiques potentielles, notamment un régime cétogène (essentiellement un régime très très pauvre en glucides). Il existe également un dispositif appelé « stimulateur du nerf vague » qui peut aider certaines personnes.
Traitement des troubles du mouvement
Il existe également des médicaments qui peuvent aider à traiter les dyskinésies paroxystiques et autres mouvements anormaux. Certains médicaments (par exemple, les benzodiazépines et l'acétazolamide) se sont déjà révélés utiles pour certaines personnes atteintes de certains troubles du mouvement qui ressemblent à ceux de KCNMA1. On n'a pas encore déterminé si ces médicaments ou d'autres médicaments pourraient aider les personnes atteintes de troubles du mouvement liés à KCNMA1.
Repos et régularité
En général, le manque de sommeil et le stress peuvent augmenter le risque ou le nombre de crises convulsives ou d’accès de mouvements anormaux, quel que soit la cause. Cela semble également être le cas chez certaines personnes présentant une canalopathie liée à KCNMA1. C’est pourquoi il faut viser une routine quotidienne régulière avec un sommeil suffisant. Le « stress » (bon et mauvais) fait partie de la vie quotidienne et ne peut être complètement évité. Il est important que toutes les personnes intègrent régulièrement des moments de repos et de relaxation à la journée, et cela est particulièrement important pour les personnes souffrant d'un problème de santé.
Physiothérapie et ergothérapie et sécurité
Un ergothérapeute peut conseiller les parents sur la meilleure façon d'aider leurs enfants à stimuler leur développement et à élaborer des plans de développement individuels. Un physiothérapeute peut conseiller les parents pour limiter les conséquences des crises et de la dyskinésie. Les personnes atteintes de dyskinésies paroxystiques et de crises convulsives risquent de se blesser. Il sera conseillé à beaucoup d'entre elles de porter un casque protecteur afin de minimiser les risques de lésions cérébrales irréversibles pouvant résulter d'une chute.
Contact avec d'autres parents
Bien que chaque enfant et chaque famille soient différents, il peut être intéressant de faire partie d’un groupe de personnes aux prises avec des problèmes similaires. Nous encourageons les patients, les soignants, les professionnels de la santé et les scientifiques à s'inscrire à nos forums de discussion. Là, vous pouvez communiquer avec d'autres patients / familles traitant de la canalopathie liée à KCNMA1. Vous pouvez trouver les forums sur www.kciaf.org ou directement via ce lien: http://www.kciaf.net.
Conseil familial
Un travailleur social ou un psychologue peut donner des conseils sur la manière dont ce trouble peut impacter la vie quotidienne des familles.

QU'EST-CE QUE LA CANALOPATHIE LIÉE À KCNMA1 SIGNIFIE POUR L'AVENIR ?
Nous ne comprenons pas tout à fait la canalopathie liée à KCNMA1 car il s’agit d’une maladie relativement récente, et peu de personnes sont connues pour en être atteintes. Cependant, on peut s’attendre à ce que le pronostic ou les résultats à long terme soient similaires à ceux d’autres crises convulsives et dyskinésiques. Le patient le plus âgé qui porte la mutation KNCMA1 est dans la cinquantaine, ce qui suggère que la durée de vie est normale. Les souris de laboratoire portant des mutations dans KCNMA1 peuvent avoir une espérance de vie normale, même si elles présentent des symptômes de problèmes cérébraux et musculaires.
RECHERCHE EN COURS
Des recherches sont en cours pour comprendre comment les mutations du gène KCNMA1 modifient l'activité des canaux potassiques qu'il code. Les recherches à venir visent à comprendre comment l'activité cérébrale peut changer avec ces mutations. Si les patients ou les familles souhaitent contribuer à cet effort de recherche, veuillez nous envoyer un courrier électronique à l'adresse www.kciaf.org et nous vous fournirons de plus amples informations.
QU'EN EST-IL DE LA MÉDECINE DE PRÉCISION ET DES THÉRAPIES QUI POURRAIMENT CORRIGER CERTAINES DÉFAUTS DU GÈNE KCNMA1 OU LA FONCTION ANORMALE DE LA PROTÉINE DE CANAL?
À l'heure actuelle, toutes les mutations de KCNMA1 n'ont pas été étudiées au niveau des cellules neuronales ou musculaires. Par conséquent, il n’est pas possible pour le moment de choisir un médicament capable de corriger spécifiquement le défaut KCNMA1. Plusieurs efforts de recherche sont en cours pour trouver de nouveaux médicaments susceptibles de compenser les symptômes de la canalopathie liée à KCNMA1, mais les recommandations actuelles sont de travailler avec des neurologues et des spécialistes des troubles du mouvement utilisant des médicaments connus et validés en clinique (effets désirables modérés et connus). Au fur et à mesure que la recherche avance, KCIAF communiquera toute nouvelle connaissance à la communauté des patients.
[An initial version of this text and figures were taken from this page, written in the Dutch language by Dr. Jolanda Scheiving, a pediatric neurologist working at the Amalia Children’s Hospital. Dr Scheiving graciously allowed us to modify and expand it for use by KCIAF.org.
Note also that this page is intended to be only general information and education on KCNMA1 disorders, and is not intended to suggest any particular treatment or specific recommendations for any particular person.